


615 - REVISIÓN DE PACIENTES AFECTOS DE ENFERMEDAD DE FABRY (EF) EN TRATAMIENTO CON TERAPIA DE SUSTITUCIÓN ENZIMÁTICA (TSE), CHAPERONAS (MIGALASTAT) Y TRATAMIENTO EXCLUSIVO SINTOMÁTICO EN UN HOSPITAL DE TERCER NIVEL
Hospital Universitario Son Espases, Palma de Mallorca.
Objetivos: Valorar características clínicas y epidemiológicas de los pacientes con enfermedad de Fabry. Determinar las mutaciones responsables y el tratamiento indicado en cada caso.
Métodos: Estudio descriptivo, observacional, retrospectivo de una serie de 10 pacientes con EF controlados en consultas de Medicina Interna en el Hospital Universitario de Son Espases desde 2004 hasta la actualidad, que se encuentran en tratamiento específico con TSE versus terapia con chaperonas o en tratamiento sintomático exclusivo. Se recogieron datos epidemiológicos, clínicos y fenotípicos, así como resultados de las pruebas complementarias realizadas y tratamientos administrados, de todos los pacientes incluidos en el estudio.
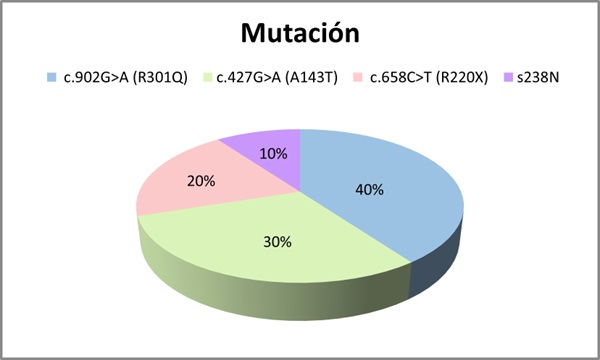
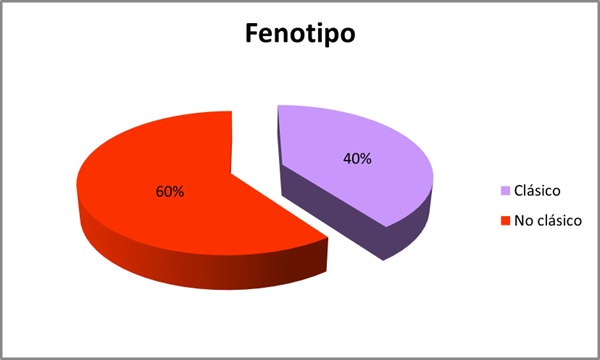
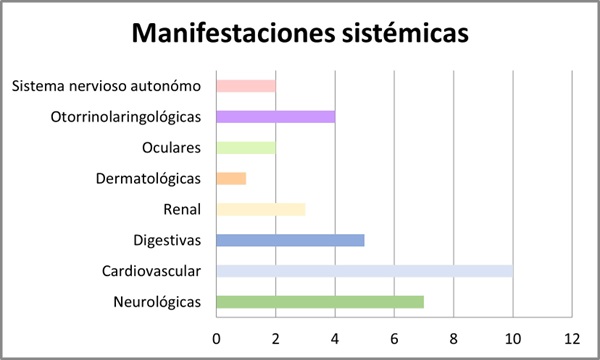
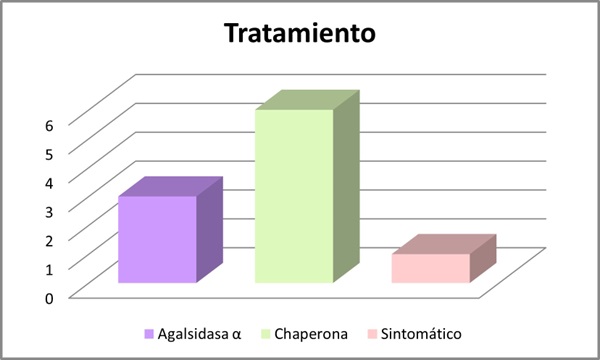
Resultados: Se incluyeron 10 pacientes, 7 hombres y 3 mujeres con una media de edad de 54 años (DE 19,697). Se analizaron las mutaciones del gen GLA: 4 pacientes con la mutación c.902G>A (R301Q), 3 con mutación c.427G>A (A143T), 2 con mutación c.658C>T (R220X) y 1 paciente s.238N (fig. 1). Se evaluó el fenotipo de cada paciente: 40% presentaban un fenotipo clásico y 60% fenotipo no clásico (fig. 2). El 70% presentaron manifestaciones neurológicas (acroparestesias), el 100% alteraciones cardiovasculares (10 presentaban hipertrofia ventricular izquierda (HVI), 5 insuficiencia cardíaca (IC), 4 arritmias y 3 ictus isquémicos), el 50% clínica digestiva, 10% angioqueratomas, 30% insuficiencia renal crónica (ERC) y proteinuria, 40% hipoacusia, 20% córnea verticillata y 20% hipohidrosis (fig. 3). En cuanto al tratamiento, el 30% recibieron TSE (agalsidasa alfa), 60% tratamiento con chaperona y 10% no recibió tratamiento por edad y situación basal (fig. 4). El 80% se encuentran estables (afectación orgánica) con el tratamiento instaurado. Una paciente fue exitus a los 82 años por ICC refractaria y 1 paciente en TSE con fenotipo clásico requirió trasplante renal por ERC estadio 5.
Discusión: La EF es una enfermedad de depósito lisosomal ligada al cromosoma X, causada por mutaciones en el gen GLA que codifica la enzima lisosomal α-galactosidasa A (α-Gal A). Las mutaciones del gen GLA reducen o anulan la actividad de α-Gal A, lo que da lugar a la acumulación progresiva de Gb3 y liso-Gb3 en el organismo produciendo disfunción orgánica. La EF es clínicamente heterogénea y progresiva, puede presentarse con fenotipo clásico o no clásico (de inicio tardío). Los pacientes con EF clásica presentan signos y síntomas característicos como dolor neuropático, córnea verticillata y angioqueratomas. La EF clásica a largo plazo y la EF de inicio tardío se manifiestan en adultos con enfermedad renal crónica (ERC), miocardiopatía hipertrófica, trastornos del ritmo cardíaco e ictus. En la EF de inicio tardío las manifestaciones pueden limitarse a un solo órgano (renal, cardíaco). La sospecha clínica de EF debe plantearse en los pacientes que presenten: HVI de etiología desconocida, ERC de etiología no filiada, ictus criptogénico en adultos jóvenes, angioqueratomas, acroparestesias, córnea verticillata y síntomas digestivos de etiología desconocida. La principal prueba diagnóstica es determinar la actividad de la enzima α-Gal A y la secuenciación del gen GLA. El tratamiento específico disponible para la EF se basa en el TSE y en la terapia con chaperonas aunque existen otros tratamientos en fase experimental.
Conclusiones: La EF es una Enfermedad Minoritaria e infradiagnosticada con manifestaciones clínicas inespecíficas que conllevan un retraso diagnóstico-terapéutico. Un 80% de nuestros pacientes en tratamiento se han estabilizado en cuanto a la afectación de los órganos diana (HVI, proteinuria). Todos nuestros pacientes en tratamiento con chaperona presentaban mutaciones genéticas susceptibles. Destacar la importancia de realizar un abordaje multidisciplinar y la necesidad de cribaje clínico y genético de los familiares. El diagnóstico e inicio de tratamiento precoz ayuda a prevenir la progresión al daño tisular irreversible.


