

Introducción
La trombosis a nivel de una placa aterosclerótica en la que se ha producido rotura o erosión constituye el sustrato fisiopatológico responsable de los síndromes coronarios agudos (SCA): infarto agudo de miocardio con y sin elevación del segmento ST y angina inestable, principales causas de mortalidad en España.
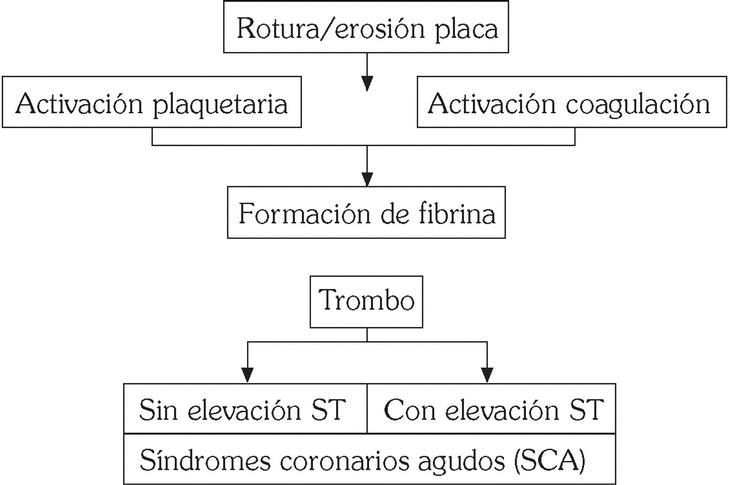
Los SCA se producen como resultado de estímulos procoagulantes actuando sobre lesiones ateroscleróticas complejas que favorecen la activación de las plaquetas y de los mecanismos de coagulación (fig. 1). La interacción entre ambos sistemas será clave en la formación del trombo en el árbol coronario 1.
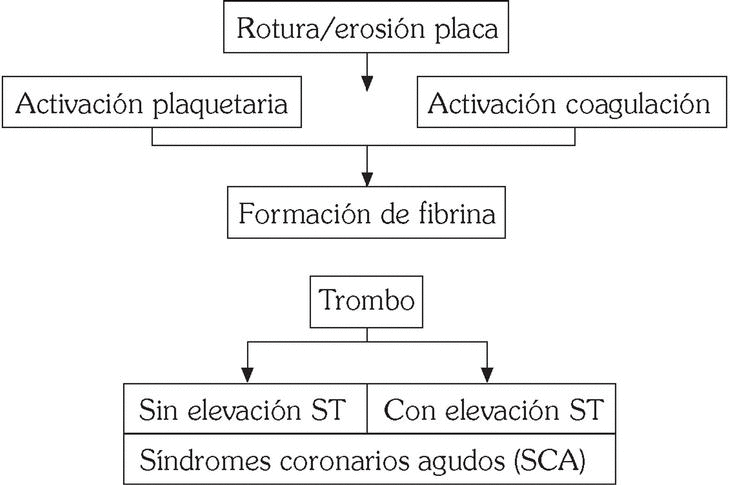
Fig. 1. Síndromes coronarios agudos: patogenia.
En ausencia de lesión vascular, cuando el endotelio está preservado, actúan sistemas endógenos de defensa antitrombóticos para mantener la permeabilidad vascular: antitrombina, proteína C/trombomodulina, inhibidor de la vía del factor tisular (TFPI) y sistema fibrinolítico. Sin embargo, en un vaso coronario aterosclerótico predominan la hipercoagulabilidad y la reducción de la actividad fibrinolítica, que favorecen la formación del trombo en las regiones más vulnerables de la placa de ateroma 2. Teniendo en cuenta el papel central de la trombosis en la patogenia de los SCA, se puede deducir la relevancia de estrategias antitrombóticas encaminadas a su prevención y tratamiento.
Mecanismos de defensa antitrombóticos: papel del endotelio vascular
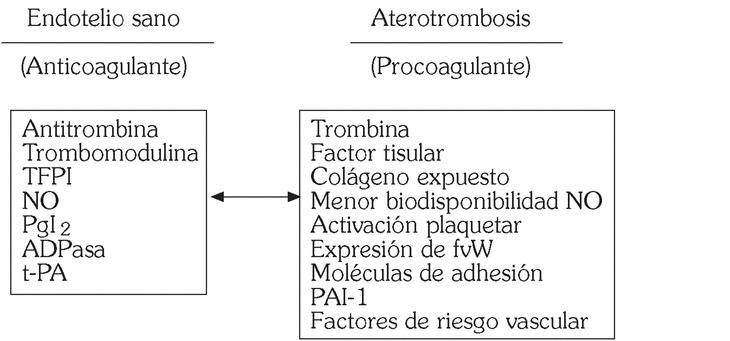
En condiciones fisiológicas el endotelio vascular es una superficie tromborresistente 3, siendo varios los mecanismos implicados (fig. 2).
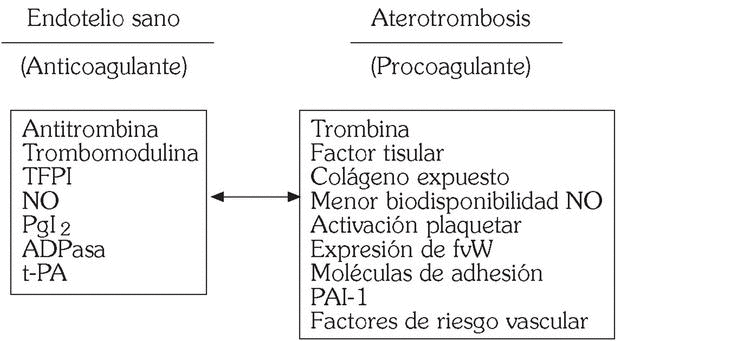
Fig. 2. Endotelio vascular: tromborresistencia frente a trombogenicidad.
Antitrombina
Es una glucoproteína sintetizada en el hígado que inhibe diversas proteasas de la coagulación, fundamentalmente trombina y factores IXa, Xa, XIa y XIIa. Dicha inhibición se acelera enormemente en presencia de moléculas de heparina y heparinoides presentes en la pared vascular, que inducen un cambio conformacional en la molécula de antitrombina, incrementado así su capacidad para neutralizar la trombina y otros factores de la coagulación.
Proteína C/trombomodulina
La proteína C es una glucoproteína vitamino-K dependiente sintetizada en el hígado que en su forma activa inhibe los factores V y VIII de la coagulación. Dicha activación ocurre a nivel de la superficie endotelial por acción del complejo formado por la trombina y un receptor endotelial específico: la trombomodulina. La proteína S, otra proteína vitamino-K dependiente expresada, asimismo, en células endoteliales, actúa como cofactor de la proteína C en la inactivación de los factores Va y VIIIa 4.
Inhibidor de la vía extrínseca (TFPI)
Es una proteína liberada por las células endoteliales y plaquetas que circula unida a lipoproteínas plasmáticas e inhibe el efecto procoagulante del complejo factor tisular/factor VII sobre los factores IXa y Xa 5.
Sistema fibrinolítico
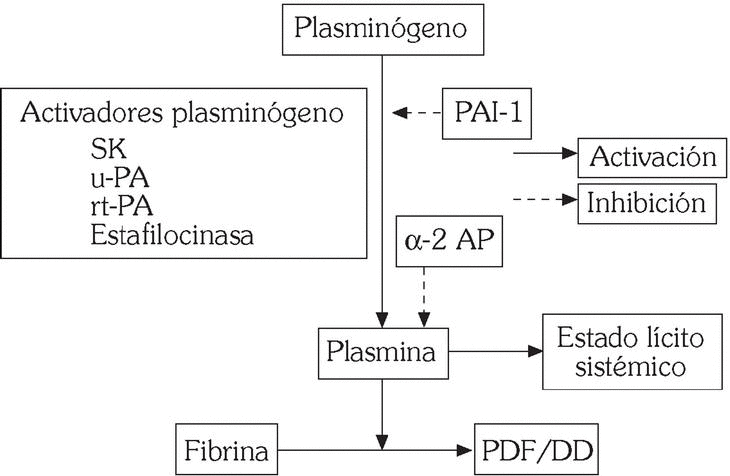
Es el encargado de la degradación enzimática de la fibrina que se realiza tras conversión de la proenzima circulante plasminógeno en la enzima activa plasmina por acción de los activadores del plasminógeno: tipo tisular (t-PA) y tipo urocinasa (u-PA). En ausencia de fibrina dicha conversión es lenta, mientras que, cuando está presente, la unión del t-PA a la fibrina acelera extraordinariamente el proceso. A su vez la degradación de la fibrina expone nuevos lugares de unión para el t-PA, lo que incrementa la lisis acelerada del coágulo 4. El sistema fibrinolítico está regulado por la acción de inhibidores específicos de la activación del plasminógeno (PAI-1) y de la plasmina (α2-antiplasmina) (fig. 3). Nuestro grupo ha demostrado un papel relevante del PAI-1 en pacientes con enfermedad coronaria 6,7. Más recientemente se ha descrito otro inhibidor de la fibrinólisis que es activado por trombina (TAFI). Se trata de una carboxipeptidasa que excinde residuos de lisina carboxiterminales en la fibrina, lo que conlleva una menor activación de la plasmina, así como la aparición de lugares de unión para el t-PA 8. Estudios preliminares sugieren un aumento significativo de los niveles de TAFI en pacientes con enfermedad coronaria.
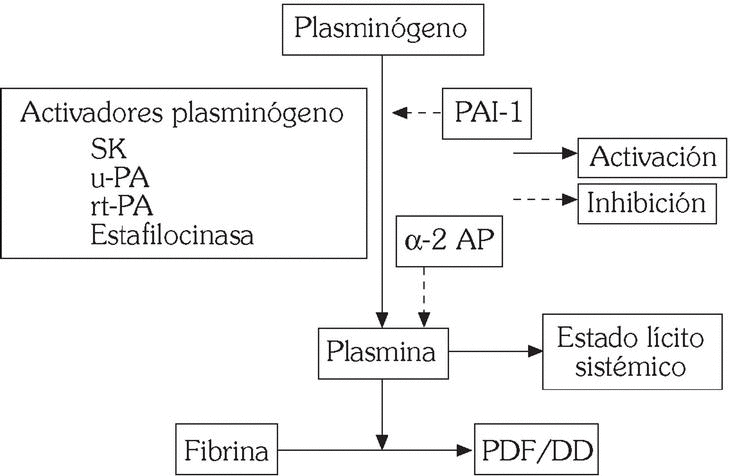
Fig. 3. Mecanismo de acción de los fármacos fibrinolíticos.
Óxido nítrico
El endotelio vascular es la fuente principal de óxido nítrico (NO), si bien las plaquetas contienen también la enzima NO-sintasa (NOS) que favorece su síntesis. Dadas las propiedades vasorrelajantes, antiproliferativas y antiagregantes plaquetarias del NO, una menor biodisponibilidad de la molécula, tal como se observa en SCA, podría constituir uno de los mecanismos implicados en el desarrollo de la trombosis. El descenso de NO en pacientes con enfermedad coronaria podría deberse a una disminución de NOS o de S-nitrosotioles, que representan reservorios endógenos de NO 9,10.
Plaquetas
En condiciones fisiológicas el endotelio vascular libera NO, prostaciclina (PGI2) y enzimas como ADPasa, que previenen la adhesión y agregación plaquetaria. Megacaciocitos y plaquetas contienen la enzima NOS encargada de la síntesis de NO 9.
Mecanismos protrombóticos: activación de las plaquetas y coagulación
Un estado procoagulante está presente en todas las etapas de la aterosclerosis, desde las fases iniciales a las de ruptura y trombosis. La activación hemostática es un fenómeno que tiene lugar sobre superficies celulares, como sucede en la aterosclerosis, donde la lesión vascular promueve los fenómenos de activación plaquetaria y de los mecanismos de coagulación 11.
Activación plaquetaria
Diversos factores de riesgo aterosclerótico (dislipidemia, tabaquismo, diabetes mellitus, hipertensión arterial) alteran las propiedades tromborresistentes del endotelio vascular favoreciendo la adhesión y la agregación de las plaquetas (fig. 2). La lesión vascular expone componentes del subendotelio, tales como colágeno y factor von Willebrand (FvW), que promueven la adhesión plaquetaria. En condiciones de altas fuerzas de cizallamiento (shear stress), características de las estenosis coronarias, la interacción del FvW con el complejo glucoproteico de membrana plaquetaria (GpIb/IX-V) promueve la adhesión de las plaquetas al colágeno, un proceso facilitado por otros receptores tales como GpIa, Ib, y VI. Diversos agonistas, como epinefrina, trombina, adenosina difosfato (ADP), colágeno, así como estímulos mecánicos, activan las plaquetas adherentes con expresión de la glucoproteína IIb/IIIa, que posee gran afinidad por el fibrinógeno 12. Esta fase conlleva el reclutamiento de plaquetas próximas y su ulterior agregación mediada por el fibrinógeno, con formación de un agregado plaquetario. Las plaquetas activadas liberan moléculas biológicamente activas que promueven la formación del trombo plaquetario y contribuyen al crecimiento de la placa aterosclerótica 13. Entre estas sustancias destacan el tromboxano A2 (TXA2), serotonina, ADP y epinefrina, que estimulan los procesos de activación y agregación plaquetaria. Además, las plaquetas liberan factores de crecimiento tales como factor de crecimiento fibroblástico (FGF), factor de crecimiento derivado de las plaquetas (PDGF), factor de crecimiento epidérmico (EGF), factor de crecimiento similar a la insulina (IGF), factor de crecimiento transformador-β (TGF-β), que estimularán la prolifera-ción de células musculares lisas, así como numerosas sustancias con propiedades proinflamatorias, como selectina P, factor-4 plaquetario, anión superóxido y ácido eicosatetranoico 14.
Por consiguiente, las plaquetas circulantes pueden ser activadas por factores de riesgo vascular y otros estímulos mecánicos, y utilizan diversas vías para interaccionar en las zonas más vulnerables de la placa de ateroma, donde promueven un ambiente trombogénico.
Activación de la coagulación: papel de la trombina y del factor tisular
La coagulación sanguínea comprende una serie de reacciones secuenciales en las que zimógenos inactivos son convertidos en enzimas activas. La etapa final consiste en la conversión del fibrinógeno en monómeros de fibrina que son polimerizados a través del factor XIIIa para formar un coágulo estable 15.
La coagulación se inicia in vivo tras la liberación del factor tisular (FT) en respuesta a la lesión vascular, clásicamente considerada como la vía extrínseca de la coagulación. Se ha demostrado la presencia de FT en el 43% de las placas ateroscleróticas de pacientes con SCA inestables y sólo en un 12% de los SCA estables 16. El FT forma un complejo con el FVIIa que es capaz de activar los factores IX y X. El factor Xa (en complejo con el Va) transforma la protrombina (factor II) en trombina (factor IIa), la cual es una enzima fundamental en la cascada de la coagulación, ya que convierte el fibrinógeno en fibrina. Además, la trombina ejerce otras acciones hemostáticas:
1) Conversión del FVIII en VIIIa y del V en Va, cofactores esenciales en la generación de trombina.
2) Conversión del FXIII en XIIIa que estabilizará la fibrina.
3) Activación plaquetaria 15.
Por otra parte, la trombina además de proagregante y procoagulante es un potente factor quimiotáctico y mitogénico para células musculares lisas 17, estimulando la producción de interleucina-6 y del factor quimiotáctico para monocitos (MCP-1). Estudios recientes implican, asimismo, a una familia de receptores activados por proteasas (PAR) como mediadores de múltiples efectos de la trombina 17,18.
La cantidad de trombina generada inicialmente es insuficiente para la formación de un coágulo estable de fibrina. Sin embargo, existen mecanismos de retro-alimentación que amplifican la cascada de la coagulación, siendo los más importantes la formación de complejos IXa/VIIIa y Xa/Va sobre la superficie de las plaquetas activadas y la activación de los factores XII y XI, clásicamente considerada como la vía intrínseca de la coagulación 15.
Por consiguiente, la aterosclerosis representaría un sustrato idóneo para la trombogénesis, ya que a la pérdida de las propiedades antitrombóticas del endotelio se sumaría un mayor potencial trombogénico que favorece el depósito de fibrina. La trombosis va a suponer, además, un estímulo adicional para el crecimiento de la placa de ateroma.
Terapéutica antitrombótica en los síndromes coronarios agudos
Terapia fibrinolítica
Los fármacos fibrinolíticos o trombolíticos promueven la conversión del plasminógeno en plasmina (fig. 3). Dado que esta enzima proteolítica posee, relativamente, escasa especificidad por la fibrina, ya que degrada otras proteínas plasmáticas como el fibrinógeno, el empleo de estos agentes conlleva un estado lítico sistémico que puede predisponer a complicaciones hemorrágicas graves 19.
Los fármacos trombolíticos se clasifican en aquellos con escasa especificidad por la fibrina (estreptocinasa, prourocinasa, urocinasa) y los que poseen mayor especificidad por ésta, tales como el t-PA recombinante (rt-PA, alteplase) y variantes del t-PA con mayor vida media, tales como reteplase, lanoteplase y TNK-t-PA (fig. 3). No obstante, el significado clínico de la especificidad por la fibrina no se ha establecido con precisión 19,20.
Si bien los agentes fibrinolíticos desempeñan un papel importante en el tratamiento precoz de los SCA con elevación ST, nivel de evidencia IA en las guías europea, americana y española 21-23, no están indicados en los restantes SCA. Finalmente, estudios recientes han demostrado que la angioplastia coronaria puede mejorar los resultados de la trombólisis en términos de reducción de la mortalidad en sujetos con infarto agudo de miocardio (IAM) y elevación del segmento ST 24,25.
Agentes antiplaquetarios
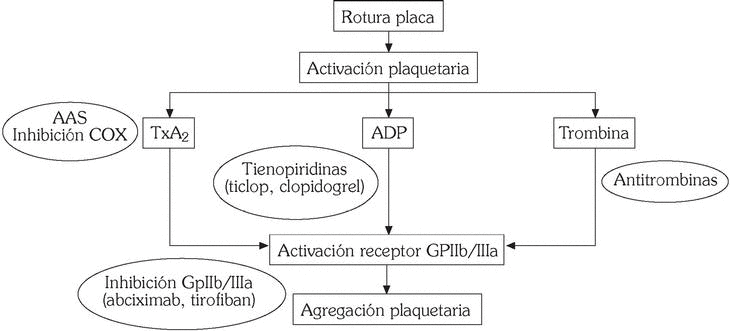
Los principales fármacos inhibidores de la función plaquetar son aspirina y derivados, tienopiridinas y antagonistas GpIIb/IIIa (fig. 4).
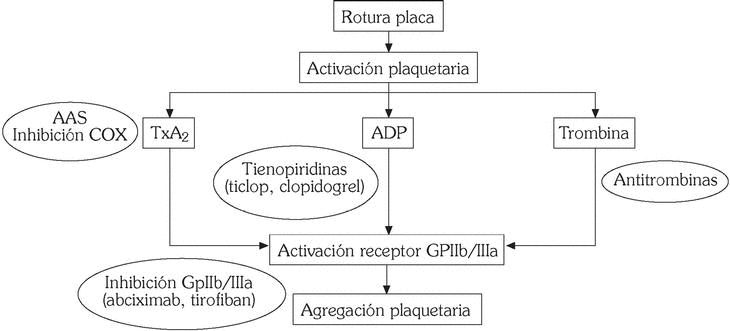
Fig. 4. Mecanismo de acción de los fármacos antiplaquetarios.
Aspirina
Inhibe irreversiblemente la enzima ciclooxigenasa-1 (COX-1), que cataliza la síntesis de TXA2 a partir del ácido araquidónico, siendo su vida media en la circulación de 20 minutos. Si bien se considera que es un débil agente antiagregante plaquetario, ya que no interfiere la activación plaquetaria por trombina y colágeno, son numerosos los estudios y metaanálisis que demuestran de forma incontestable un claro efecto beneficioso en el tratamiento de los SCA 26,27.
Tienopiridinas (ticlopidina, clopidogrel)
Inhiben la agregación plaquetaria inducida por ADP, así como la activación plaquetaria en respuesta a otros estímulos, como factor activador plaquetario y colágeno. En cuanto a su mecanismo de acción requieren la conversión a un metabolito activo y la unión a un receptor específico para el ADP 28.
La descripción de neutropenias, púrpura trombótica trombocitopénica y aplasia medular grave ha limitado el uso de ticlopidina. Sin embargo, el clopidogrel ofrece un perfil farmacocinético y de seguridad más favorable, por lo que su uso se ha generalizado en SCA con y sin elevación ST, sometidos o no a angioplastia coronaria 29. En un amplio estudio clínico, el PCI-CURE, se ha demostrado la eficacia de la combinación clopidogrel/aspirina pre y postangioplastia, si bien se observó un aumento significativo de las complicaciones hemorrágicas 30.
Antagonistas glucoproteína plaquetaria IIb/IIIa
Teniendo en cuenta que la GpIIb/IIIa representa la vía final común a través de la cual actúan todos los agonistas plaquetarios, la neutralización de este receptor ha supuesto un importante avance en el tratamiento de los SCA. Se dispone en la actualidad de diversos preparados por vía intravenosa: abciximab (Reopro), eptifibatide (Integrelin), tirofiban (Aggrastat). Si bien estos agentes poseen diferente estructura química y perfil farmacocinético, son diversos los estudios que han demostrado una reducción significativa en la recurrencia de episodios cardiovasculares, así como de la necesidad de cirugía de revascularización coronaria en pacientes con SCA que son sometidos a estrategias de intervencionismo vascular (angioplastia y/o stent), por lo que su uso se ha gene- ralizado en las unidades de hemodinámica y coronarias 31,32. Entre sus efectos adversos destacan aumento de complicaciones hemorrágicas y trombocitopenia.
Es preciso señalar que estudios que han empleado antagonistas IIb/IIIa por vía oral no han podido demostrar ningún efecto beneficioso, e incluso se han observado mayores tasas de mortalidad 33.
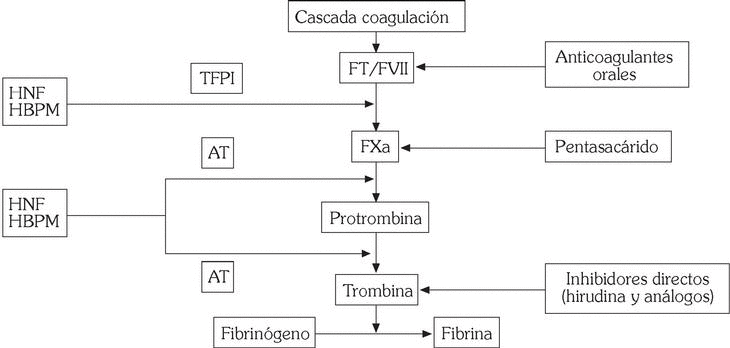
Inhibidores de la trombina
Se incluyen en este apartado inhibidores indirectos de la trombina, como heparina no fraccionada (HNF), de bajo peso molecular (HBPM) e inhibidores del factor X (pentasacárido), y sustancias que interfieren directamente con la trombina, como hirudina y análogos (fig. 5).
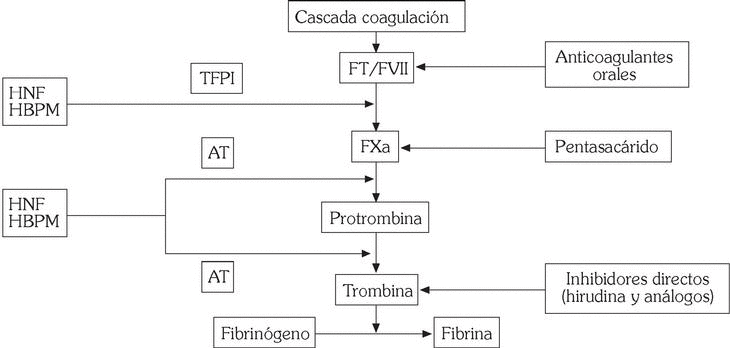
Fig. 5. Mecanismo de acción de los fármacos anticoagulantes.
HNF, HBPM y pentasacárido
Si bien el tratamiento estándar con anticoagulantes en la fase aguda de los SCA ha consistido en la administración de HNF por vía intravenosa, en la actualidad se prefiere el empleo de HBPM, que se obtienen a partir de la HNF mediante polimerización enzimática o química. Se diferencian de ésta por su elevada actividad anti-Xa en relación a su efecto antitrombina, mayor biodisponibilidad y vida media; no necesitan monitorización y se administran por vía subcutánea, por lo que pueden emplearse de forma ambulatoria 34. Son diversos los preparados disponibles (ardeparina, dalteparina, enoxaparina, fraxiparina, reviparina, tinzaparina), pero la enoxaparina ha sido el fármaco que ha demostrado una mayor reducción de episodios isquémicos en los SCA en comparación con otros preparados 35-38. En pacientes sometidos a intervencionismo coronario, tanto dalte- parina como enoxaparina, en combinación con antagonistas IIb/IIIa, se han mostrado eficaces y la asociación no aumenta el riesgo de complicaciones hemorrágicas graves 39-41. Asimismo, algunos estudios combinando enoxaparina con agentes fibrinolíticos han demostrado una mejoría en la permeabilidad coronaria 42,43. Sin embargo, se precisan estudios clínicos randomizados que identifiquen qué subgrupos de pacientes con enfermedad arterial coronaria pueden beneficiarse en mayor medida del empleo de HBPM.
Fondaparinux, un pentasacárido sintético, es el primero de una nueva clase de fármacos antitrombóticos que inhiben de forma selectiva el factor Xa y que muestra una gran eficacia en la prevención del tromboembolismo venoso tras cirugía ortopédica 44. Estudios recientes, como el PENTALYSE, también han demostrado que la administración de este fármaco por vía subcutánea es al menos tan segura y efectiva como la HNF en el manejo de pacientes con SCA 45.
Hirudina y análogos
Son polipéptidos que inactivan tanto la trombina libre como la unida a la fibrina, por lo que, desde un punto de vista teórico, estarían indicados en pacientes con SCA. Se han desarrollado diversos preparados: hirudina, bivalirudina, argatroban, inogatran, efegatran. Estudios preliminares sugieren que bivalirudina e hirudina reducen las complicaciones isquémicas en relación con HNF, si bien este efecto no fue significativo en la valoración a largo plazo y se asoció con un incremento de complicaciones hemorrágicas 46,47. Estudios en curso delimitarán el papel de inhibidores orales de trombina (ximelagatran) en SCA.
Otros fármacos con propiedades antitrombóticas
Las estatinas, además de su efecto hipolipidemiante, ejercen numerosas acciones (efectos pleiotrópicos), entre las cuales destacan acciones antitrombóticas, como la disminución de factores protrombóticos (FT y PAI-1) y antiinflamatorias (disminución de proteína C reactiva), por lo que se han ido incorporando en el manejo de los pacientes con enfermedad arterial coronaria, incluso durante la fase aguda 48,49.
Los inhibidores de la enzima conversora de la angiotensina (IECA) como ramipril, administrado pre y postrombólisis reducen la incidencia de episodios isquémicos y mejoran el balance fibrinolítico en pacientes con SCA 50.
Conclusiones
En la actualidad se conocen con más precisión los mecanismos celulares y humorales que subyacen en la patogenia de los SCA, favoreciendo la activación hemostática responsable de la trombosis. El factor tisular se considera clave en el desarrollo del proceso trombótico responsable de la isquemia y necrosis miocárdica. La trombosis va a ser, asimismo, relevante en el crecimiento ulterior de la placa de ateroma. Numerosos factores tradicionales de riesgo vascular suponen un potente estímulo trombogénico, por lo que el tratamiento agresivo de dichos factores continúa siendo clave en la prevención de los SCA.
La aspirina es un antiagregante muy eficaz en pacientes con SCA, pero incapaz de neutralizar el efecto de los numerosos agonistas plaquetarios presentes en la placa de ateroma, por lo que debería emplearse en combinación con otros antiplaquetarios, sobre todo en pacientes sometidos a angioplastia coronaria. Por otra parte es cada vez mayor la evidencia de que las HBPM pueden sustituir a la HNF en la mayoría de los pacientes con SCA, incluyendo aquellos sometidos a intervencionismo vascular, donde el empleo de antagonistas IIb/IIIa se asocia con una reducción significativa de la mortalidad y recurrencia de episodios cardiovasculares. Finalmente, el clopidogrel solo o combinado con aspirina emerge como un fármaco muy prometedor en el tratamiento a corto y largo plazo de pacientes con SCA, incluso en aquellos en los que se plantea la realización de angioplastia o colocación de un stent intravascular.
Un mayor conocimiento de los mecanismos básicos implicados en la patogenia de la trombosis permitirá el futuro desarrollo de nuevas estrategias antitrombóticas, con mayor especificidad de acción y menores efectos adversos.


