


742 - CARACTERÍSTICAS CLÍNICO-EPIDEMIOLÓGICAS DE LA COHORTE DE PACIENTES CON TELANGIECTASIA HEMORRÁGICA HEREDITARIA (HHT) O SÍNDROME DE RENDU-OSLER-WEBER, EN SEGUIMIENTO EN UN HOSPITAL DE TERCER NIVEL
Hospital Universitario Virgen de las Nieves, Granada, España.
Objetivos: La telangiectasia hemorrágica hereditaria, también llamada síndrome de Rendu-Osler-Weber, es un trastorno vascular hereditario con un rasgo autosómico dominante con penetrancia y expresión variables, presentando gran variedad de manifestaciones clínicas, incluso entre familiares que tienen la misma variante del gen patogénico. El objetivo del presente trabajo es describir las características clínico-epidemiológicas y comorbilidades de una cohorte de pacientes con HTT en seguimiento en consulta de Enfermedades Minoritarias en un hospital universitario de tercer nivel.
Métodos: Se trata de un estudio observacional, unicéntrico y descriptivo, donde se recogieron retrospectivamente, mediante revisión de las historias clínicas, variables clínico-epidemiológicas de los pacientes con HHT en seguimiento activo en una consulta específica de Enfermedades Minoritarias perteneciente a Medicina Interna del Hospital Universitario Virgen de las Nieves (Granada). Para su realización, se obtuvieron los consentimientos requeridos y se contó con la aprobación del Comité Ético.
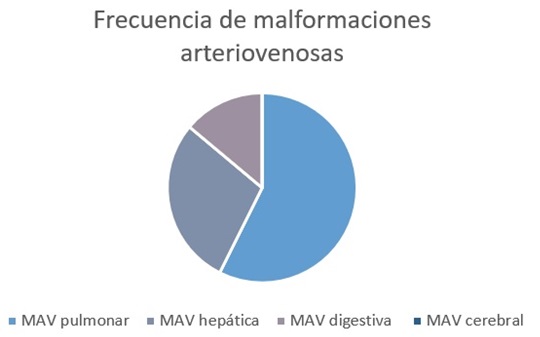
Resultados: Se recopilaron los datos de un total de 6 pacientes con criterios diagnósticos de HTT o síndrome de Rendu-Osler-Weber, 2 mujeres (33,3%) y 4 hombres (66,7%), con una edad media de 50,83 años (DT ± 13,8 años) y una edad media al diagnóstico de 48,67 años (DT ± 13,53 años). De los cuales, 3 pacientes fueron diagnosticados de HHT tipo 1 (pérdida de función en ENG) y otros 3 de HTT tipo 2 (pérdida de función en ACVRL1). 3 pacientes (50%), presentaban antecedentes familiares de primer grado de enfermedad. El resto, fueron caso índice. Las manifestaciones clínicas más frecuentes fueron la epistaxis de repetición y las telangiectasias mucocutáneas. Todos los pacientes, presentaban telangiectasias mucocutáneas al diagnóstico. El 83,3% de los pacientes tuvieron algún episodio de epistaxis a lo largo de su vida, requiriendo 3 de ellos intervencionismo otorrinolaringológico en forma de taponamiento/cauterización. El 33,3% presentó historia de anemia microcítica por pérdidas insensibles, requiriendo terapia con hierro oral. Un paciente (16,7%) presentó historia de sagrado agudo gastrointestinal, con requerimiento de soporte transfusional. De la cohorte de pacientes con HTT, 4 pacientes (66,7%), presentaron malformaciones arteriovenosas (MAV) pulmonares, 2 (33,3%) presentaron MAV hepáticas, 1 (16,7%) MAV intestinales (en forma de angiodisplasia) y ninguno MAV cerebrales (fig.). Dos pacientes (33,3%) presentaron eventos cerebrales como consecuencia de embolismos procedentes de las MAV pulmonares; fueron ictus isquémico en área vertebrobasilar y absceso piógeno, respectivamente. Los 4 pacientes con MAV pulmonares fueron sometidos a embolización por parte de Radiología Intervencionista. El paciente con MAV intestinal fue sometido a termocoagulación con plasma de argón, mientras que las MAV hepáticas no pudieron embolizarse por su gran tamaño. Ningún paciente presentó eventos trombóticos, indicación de terapia con antifactor de crecimiento vascular endotelial o inmunomoduladores. Un paciente recibió tratamiento anticoagulante con edoxabán.
|
Manifestación clínica |
Frecuencia de presentación |
|
Telangiectasias mucocutáneas |
6 (100%) |
|
Epistaxis de repetición |
5 (83,3%) |
|
Hemorragia gastrointestinal aguda |
1(16,7%) |
|
Ferropenia |
2 (33,3%) |
|
MAV pulmonar |
4 (66,7%) |
|
MAV hepática |
2 (33,3%) |
|
MAV cerebral |
0 |
|
MAV digestiva |
1 (16,7%) |
|
Eventos embólicos cerebrales |
2 (33,3%) |
|
Trombosis arteriales o venosas |
0 |
Conclusiones: Observamos la heterogeneidad en la presentación clínica de la HTT, así como la necesidad de seguimiento de esta enfermedad en una unidad específica en Enfermedades Minoritarias, con experiencia en el tratamiento y seguimiento de este tipo de dolencias menos frecuentes en nuestra práctica clínica diaria.
Bibliografía
- Mejía ARR, Fuertes MY, Moya MJ. Brain Abscess in a Patient with Rendu-Osler-Weber Syndrome: Value of Proton Magnetic Resonance Spectroscopy. NMC Case Rep J. 2016;3(2):35-7.
- Boother EJ, Brownlow S, Tighe HC, et al. Cerebral Abscess Associated With Odontogenic Bacteremias, Hypoxemia, and Iron Loading in Immunocompetent Patients With Right-to-Left Shunting Through Pulmonary Arteriovenous Malformations. Clin Infect Dis. 2017 Aug 15;65(4):595-603.
- Etievant J, Si-Mohamed S, Vinurel N, et al. Pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia: Correlations between computed tomography findings and cerebral complications. Eur Radiol. 2018 Mar;28(3):1338-44.
- Corvino F, Silvestre M, Cervo A, et al. Endovascular occlusion of pulmonary arteriovenous malformations with the ArtVentive Endoluminal Occlusion System™. Diagn Interv Radiol. 2016;22(5):463-5.

